Implicit hydrogen bond interaction#
This tutorial showcases how to calculate hydrogen bond (H-bond) interations for a topology with only heavy atoms (implicit hydrogens). Also, the difference between the implicit and explicit methods are demonstrated.
Before getting started to the implicit H-bond calculation, we need to make sure the bond orders of molecules are corrected. We introduce a protein helper function (ProteinHelper) to do all in once.
Protein helper function#
The protein helper function is designed to fix the bond orders in your topology file based on a given molecule template. The algorithms of fixing the bond orders are inspired by pdbinf and RDKit’s function (rdkit.Chem.AllChem.AssignBondOrdersFromTemplate).
A small example: ACE-TPO-NME#
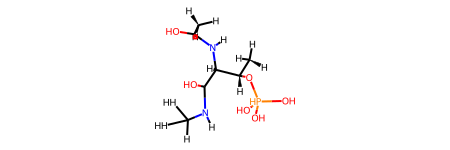
This example is originally come from pdbinf. This example shows a molecule with three residues: ACE, TPO, NME. You can find the correct bond orders at RCSB PDB.
from rdkit import Chem
from rdkit.Chem.rdDetermineBonds import DetermineConnectivity
from prolif import Molecule
from prolif.datafiles import datapath
protein_path = datapath / "protein_helper/tpo.pdb"
# Read the data with RDKit
mol = Chem.MolFromPDBFile(str(protein_path), removeHs=False)
DetermineConnectivity(mol, useHueckel=True)
for atm in mol.GetAtoms():
atm.SetNoImplicit(False) # set no implicit to False
# Convert to ProLIF Molecule
mol = Molecule.from_rdkit(mol)
print([res for res in mol.residues])
mol
/home/docs/checkouts/readthedocs.org/user_builds/prolif/envs/302/lib/python3.11/site-packages/MDAnalysis/topology/TPRParser.py:161: DeprecationWarning: 'xdrlib' is deprecated and slated for removal in Python 3.13
import xdrlib
[ResidueId(ACE, 1, ), ResidueId(TPO, 2, ), ResidueId(NME, 3, )]
import prolif as plf
plf.display_residues(mol)

Considering these three residues are not standard protein residues (20 amino acids), you will need to provide a template for the additional residues. We provide two ways to load your templates into your protein helper function.
CIF template: you can look up your residue (ligand) at RCSB PDB’s ligand page and download it by clicking Download Files > Definition (CIF format).
SMILES string template: simply provide a SMILES string for your residue or ligand.
from prolif.datafiles import datapath
from prolif.io.cif import cif_template_reader
from prolif.io.protein_helper import ProteinHelper
# load templates
ace_template = {"ACE": {"SMILES": "CC=O"}}
tpo_template = {"TPO": {"SMILES": "C[C@H]([C@@H](C(=O))N)OP(=O)(O)O"}}
# tpo_template = cif_template_reader(
# datapath / "protein_helper/templates/TPO.cif"
# ) # alternative
nme_template = cif_template_reader(datapath / "protein_helper/templates/NME.cif")
# fix molecule by the templates
protein_helper = ProteinHelper(templates=[ace_template, tpo_template, nme_template])
Important
If you would like to use SMILES templates, you have to first indicate your residue name, and then define your SMILES string in a dictionary with key SMILES. For instance:
{
"[residue name]": {
"SMILES": "[your smiles string]"
},
...
}
Tip
If using SMILES templates, you might need to modify your SMILES string by pruning one oxygen in an acetate group (especially, if your residue is not in the terminus). For instance in the above example, original TPO residue’s SMILES is C[C@H]([C@@H](C(=O)O)N)OP(=O)(O)O (if you look at RCSB TPO page), but your template should be C[C@H]([C@@H](C(=O))N)OP(=O)(O)O. It is because prolif splits your molecule into residues at the amide bonds before calculating molecular interactions. The ProteinHelper is correcting your residues, so you actually have to provide templates without -OH in the acetate group forming amide bond.
Let’s fix the residues.
# fix molecule by the templates
fixed_mol = protein_helper.standardize_protein(mol)
plf.display_residues(mol)

Now, it works.
After learning how to use protein helper function, we can now learn to calculate the implicit H-bond interactions.
Implicit H-bond interactions#
First, we read the receptor (protein) and fix the bond orders with ProteinHelper.
# read the receptor
from rdkit import Chem
from prolif.datafiles import datapath
from prolif.io.protein_helper import ProteinHelper
data_path = datapath / "implicitHbond"
protein_path_i = data_path / "receptor.pdb"
protein_mol_i = Molecule.from_rdkit(Chem.MolFromPDBFile((str(protein_path_i))))
protein_helper = ProteinHelper()
# fix the receptor
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
plf.display_residues(protein_mol_i, slice(100, 108))
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue MET1.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue LYS2.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue VAL5.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue ARG22.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue LYS27.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue GLU30.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue LYS34.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue LYS55.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue GLU58.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/home/docs/checkouts/readthedocs.org/user_builds/prolif/checkouts/302/prolif/io/protein_helper.py:161: UserWarning: Could not guess the forcefield based on the residue names. CYS is assigned to neutral CYS (charge = 0).
standardized_resname = self.convert_to_standard_resname(
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue GLU122.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue GLU124.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue LYS125.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue LYS126.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue THR143.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue GLU146.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue LYS147.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue GLU155.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue TYR167.A has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue LYS2.B has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue VAL5.B has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue SER18.B has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue LYS55.B has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue ASN56.B has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue GLU58.B has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue GLU119.B has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)
/tmp/ipykernel_1219/216093026.py:14: UserWarning: Residue GLU146.B has a different number of heavy atoms than the standard residue. This may affect H-bond detection.
protein_mol_i = protein_helper.standardize_protein(protein_mol_i)

Don’t worry if there is a lot of warning message generated. It is because your topology file is not complete for some residues (e.g., only have backbone and miss some heavy atoms at the side chain). If the residues are not close to your ligand, it should be fine.
Note
The protein helper function automatically determines the appropriate forcefield based on the residue name. This is important because, in certain forcefields, specific residue names correspond to particular protonation states of the residue. The function ensures that your residue is adjusted to match the correct protonation state associated with the selected forcefield.
In the above example, the protein helper function fails to guess the forcefield, and thus it assigns the residues’ protonated states based on default settings (basically, pH = 7 and CYS is neutralized).
Tip
You can modify the residue name for your residue to get different protonated state of the residue when using protein helper function. This approach is particularly useful when investigating different protonation states of residues.
Next, we read the ligand. Because the file is in .sdf format, we used prolif.sdf_supplier to read it.
# Read the ligand
ligand_i = plf.sdf_supplier(data_path / "1.D.sdf")[0]
plf.display_residues(ligand_i, size=(400, 200))

We then used prolif.Fingerprint to find the implicit hydrogen bonds by setting interactions= ["ImplicitHBAcceptor", "ImplicitHBDonor"].
Then, we used run_from_iterable to calculate the implicit H-bond interactions.
fp_i = plf.Fingerprint(
interactions=["ImplicitHBAcceptor", "ImplicitHBDonor"],
count=True,
)
fp_i.run_from_iterable([ligand_i], protein_mol_i)
<prolif.fingerprint.Fingerprint: 2 interactions: ['ImplicitHBAcceptor', 'ImplicitHBDonor'] at 0x71cf3fd47ad0>
Note
Some parameters for the implicit H-bond method can be set.
For instance, if you want to include waters for detecting interactions, use "include_water": True.
Geometric checks play an important role in the implicit hydrogen bond method by filtering out impossible interaction pairs. You can make the method more flexible by adjusting the tolerance thresholds for acceptor or donor atom angles and plane angles (plane angles only for sp2-hybridized heavy atoms). This allows you to include more or fewer interactions based on your needs. Increasing the tolerance reduces the geometric restrictions. Our default settings are fine-tuned by a dataset from RCSB PDB. If you don’t want any geometry check, you can simply apply "ignore_geometry_checks": True.
Here is an example to adjust the parameters:
fp_i = plf.Fingerprint(
interactions=["ImplicitHBAcceptor", "ImplicitHBDonor"],
count=True,
parameters={
"ImplicitHBAcceptor": {
"include_water": True, # include water residues in the detection
"tolerance_dev_daa": 30, # atom angle deviation tolerance for donor
"tolerance_dev_dpa": 30, # plane angle deviation tolerance for donor
"ignore_geometry_checks": True, # to skip geometry checks
},
"ImplicitHBDonor": {
"include_water": True, # include water residues in the detection
"tolerance_dev_daa": 30, # atom angle deviation tolerance for donor
"tolerance_dev_dpa": 30, # plane angle deviation tolerance for donor
"ignore_geometry_checks": False, # not to skip geometry checks
},
},
)
Similar to the previous tutorials, we converted the fingerprint into a table.
df_i = fp_i.to_dataframe().T
df_i
| Frame | 0 | ||
|---|---|---|---|
| ligand | protein | interaction | |
| UNL1 | SER14.A | ImplicitHBAcceptor | 1 |
| ImplicitHBDonor | 1 | ||
| TYR17.A | ImplicitHBAcceptor | 1 | |
| ImplicitHBDonor | 1 | ||
| ASP95.A | ImplicitHBDonor | 1 | |
| GLU101.A | ImplicitHBDonor | 2 | |
| TYR167.B | ImplicitHBDonor | 1 |
Metadata#
If you would like to have more details (metadata), use .ifp with the key ("[ligand's resname][resid].[chainid]", "[protine's resname][resid].[chainid]"). Mostly, metadata are related to the geometric checks, including raw atom and plane angles and their deviations. We also included vina_hbond_potential to assess the likelihood of an interaction occurring. Higher values of vina_hbond_potential suggest a stronger probability of the interaction.
fp_i.ifp[0][("UNL1", "TYR167.B")]
{'ImplicitHBDonor': ({'indices': {'ligand': (11,), 'protein': (3,)},
'parent_indices': {'ligand': (11,), 'protein': (2508,)},
'distance': 2.7428045865500525,
'ideal_acceptor_angle': 120.0,
'acceptor_atom_angles': [119.56582217170318],
'acceptor_atom_angle_deviation': 0.43417782829682494,
'acceptor_plane_angle': 14.191233519808888,
'ideal_donor_angle': 120.0,
'donor_atom_angles': [140.58040843212183],
'donor_atom_angle_deviation': 20.58040843212183,
'donor_plane_angle': 1.9892680854814626,
'vina_hbond_potential': 0.9921165792423713},)}
Tip
If you have underscore (_) in your chain ID, you might have KeyError. Try to use ResidueId rather than string format as the dictionary key.
>>> fp_i.ifp[0][("UNL1", "HOH1._")] # Not working
KeyError: (ResidueId(UNL, 1, None), ResidueId(HOH, 1, None))
# try this
>>> from prolif.residue import ResidueId
>>> fp_i.ifp[0][(ResidueId("UNL", 1, None), ResidueId("HOH", 1, "_"))]
{'ImplicitHBAcceptor': ... }
Visualization#
For 2D ligand-centered plot, use plot_lignetwork with your ligand.
view = fp_i.plot_lignetwork(ligand_i, kind="frame", frame=0, display_all=True)
view
For a protein-ligand complex, plot_3d is a better way to explore the interactions. It is noted that you might need to set the water molecules into "sphere" to visualize it.
view = fp_i.plot_3d(
ligand_i, protein_mol_i, frame=0, display_all=True, remove_hydrogens=False
)
view.setStyle(
{
"resn": "HOH",
},
{"sphere": {"radius": 0.5, "color": "red"}},
)
view
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
<prolif.plotting.complex3d.Complex3D at 0x71cf3feb13d0>
Explicit H-bond interaction method (standard method)#
As a reference, we also calculated the explicit H-bond interactions for the same system.
import MDAnalysis as mda
from prolif import Molecule
from prolif.datafiles import datapath
# Read the protonated protein file
data_path = datapath / "implicitHbond"
protein_path = data_path / "receptor_ph7_amber.pdb"
u = mda.Universe(str(protein_path))
protein_mol = Molecule.from_mda(u)
plf.display_residues(protein_mol, slice(100, 108))

# Read the protonated ligand file
ligand = plf.sdf_supplier(str(data_path / "1.D_protonated.sdf"))[0]
plf.display_residues(ligand, size=(400, 200))
# Calculate default H-bond interactions
fp_count = plf.Fingerprint(["HBDonor", "HBAcceptor"], count=True)
fp_count.run_from_iterable([ligand], protein_mol)
<prolif.fingerprint.Fingerprint: 2 interactions: ['HBDonor', 'HBAcceptor'] at 0x71cf3dc00f10>
# Get the fingerprint DataFrame
df = fp_count.to_dataframe().T
df
| Frame | 0 | ||
|---|---|---|---|
| ligand | protein | interaction | |
| UNL1 | SER14.A | HBAcceptor | 1 |
| TYR17.A | HBDonor | 1 | |
| TYR167.B | HBDonor | 1 |
2D/3D Visualization#
view = fp_count.plot_lignetwork(ligand, kind="frame", frame=0, display_all=True)
view
view = fp_count.plot_3d(
ligand, protein_mol, frame=0, display_all=False, remove_hydrogens=False
)
view
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
<prolif.plotting.complex3d.Complex3D at 0x71cf3d721510>
Comparison between explicit and implicit H-bond interaction methods#
Finally, let’s compare the explicit and implicit hydrogen bond interaction methods. As shown in the visualization below, the implicit method tends to detect more interactions. This is because the explicit hydrogen bond method relies on how hydrogens are added to the ligand and protein structures. For some rotatable hydrogen donors, you can imagine these donors as being flexible during molecular dynamics simulations. The explicit method typically captures a single conformational state of hydrogen bond interactions, whereas the implicit method can be seen as integrating results from multiple conformational states. This is the fundamental difference between the two methods, and neither is inherently superior. The choice depends on the type of information you aim to extract from your data.
from prolif.plotting.complex3d import Complex3D
# create Complex3D objects (explicit, left)
comp3D = Complex3D.from_fingerprint(fp_count, ligand, protein_mol, frame=0)
# (implicit, right)
other_comp3D = Complex3D.from_fingerprint(fp_i, ligand_i, protein_mol_i, frame=0)
# compare the two Complex3D objects
view = comp3D.compare(other_comp3D, display_all=True)
view
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
<prolif.plotting.complex3d.Complex3D at 0x71cf3d7ae750>